Адреногенитальный синдром (врожденная дисфункция коры надпочечников, врожденная гиперплазия коры надпочечников) — группа наследственных болезней, в основе которых лежит недостаточность ферментов на различных уровнях синтеза стероидных гормонов коры надпочечников — кортизола и альдостерона.
Тип наследования аутосомно-рецессивный. Частота 1:5000-1 :6500.
Патогенез.
Наследственный дефект в ферментативных системах (в большинстве случаев дефицит или недостаточность 21-гидроксилазы и дефицит 11-гидроксилазы; реже встречаются недостаточность 3-бета-ол-дегидрогеназы, дефицит 18- и 77-гидроксилаз, дефицит 20-22-десмолаз и др.) приводит к снижению содержания в крови кортизола и альдостерона. Синтез половых гормонов при этом в коре надпочечников не нарушается. Низкий уровень кортизола в крови по принципу обратной связи стимулирует гипоталамо-гипофизарную систему и повышение секреции АКТГ.
В свою очередь высокий уровень АКТГ способствует гиперплазии коры надпочечников именно той зоны, в которой не нарушен синтез гормонов — преимущественно андрогенов. Одновременно с андрогенами образуются промежуточные продукты синтеза кортизола.
В зависимости от характера ферментативного дефекта выделяют следующие формы АГС: вирильную (простую, компенсированную) и сольтеряющую.
Вирильная форма — наиболее частая форма синдрома; она обусловлена частичной недостаточностью 21-гидроксилаэы. При этой форме нарушается только синтез глюкокортикоидов, что частично компенсируется гиперплазией надпочечников и приводит клатентной надпочечниковой недостаточности.
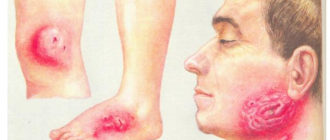
Гиперпродукция андрогенов, начинающаяся еще внутриутробно, приводит к андрогенизации вторичных половых признаков плода и рождению девочек по признаками ложного женского гермафродитизма, а мальчиков — с увеличенным половым членом. Имеет место гиперпигментация наружных половых органов, кожных складок, ареол вокруг сосков, анального отверстия. Если диагноз после рождения не поставлен, то в дальнейшем характерно появление признаков преждевременного полового созревания (в среднем в 2- 4 года), сопровождающегося маскулинизацией, ранним половым оволосением, низким голосом, acne vulgaris, ускорением роста.
Вследствие раннего закрытия зон роста дети остаются низкорослыми. Степень выраженности указанных симптомов может варьировать в довольно широких пределах.
Диагноз, помимо данных анамнеза и клиники, основывается на данных рентгенографии кистей рук (ускорение костного возраста), выявлении повышенной экскреции с мочой 17-кетостероидов (17-КС), снижения экскреции 17-оксикортикостерои-дов, высокого уровня в крови АКТГ, 17-оксипрогестерона.
Дифференциальный диагноз проводят с надпочечниковой недостаточностью, гермафродитизмом другого генеза, различными вариантами преждевременного полового созревания, андрогенпродуцирующей опухолью надпочечников.
Лечение. Глюкокортикоиды пожизненно. Дозу подбирают индивидуально под контролем 17-КС в суточной моче. Психотерапия. При необходимости проводят пластику наружных половых органов — пластику влагалища, клиторэктомию. Прогноз при своевременно начатом лечении для жизни благоприятный.
Сольтеряющая форма (более редкая, обусловлена полным блоком 21-гидроксилазы). При этой форме нарушается синтез не только глюкокортикоидов (гидрокортизона, кортизона), но и минералокортикоидов (альдостерона), что ведет, помимо андрогенизации, к усиленному выводу из организма натрия и хлоридов и к гиперкалиемии.
Наиболее ранними симптомами, кроме андрогенизации, являются отмечающиеся с рождения рвота фонтаном, как правило, не связанная с приемом пищи, жидкий стул. Развивается эксикоз, возможны судороги. Прогрессирующее нарушение водно-солевого баланса заканчивается коллапсом и расстройством сердечного ритма, а затем наступает летальный исход. Клиническая картина при этой форме напоминает пилоростеноз (псевдопилоростеноз). Диагноз основывается на тех же критериях, что и при вирильной форме.
Дифференциальный диагноз, помимо заболеваний, указанных при вирильной форме, проводится с пилоростенозом, кишечными инфекциями, токсическим синдромом.
Лечение. Используют глюкокортикоиды, как и при вирильной форме, но в сочетании с минералокортикоидами (дезоксикортикостерона ацетат-ДОКСА).
Прогноз при своевременно начатом лечении относительно благоприятный.
Гипертоническая форма — наиболее редкая, обусловлена дефицитом 11-гидроксилазы, в результате чего, как и при вирильной форме, снижается синтез кортизола и увеличивается продукция андрогенов. По пути синтеза минералокортикоидов снижается образование альдостерона, но в повышенных количествах накапливается 11-дезоксикортикостерон (у здоровых расщепляющийся 11-гидроксилазой). Он обладает минералокортикоидными свойствами и способствует задержке натрия в организме, что обусловливает длительную артериальную гипертензию, осложняющуюся кровоизлияниями в мозг с развитием гемипареза, декомпенсацией сердечной деятельности, изменением глазного дна, сосудов почек и др. Манифестация процесса наступает после З лет, но бывает и более раннее начало.
Диагностика и дифференциальная диагностика те же, что и при вирильной форме, но с учетом артериальной гипертензии.
Лечение то же, что и при вирильной форме.
Прогноз для жизни при своевременно начатом лечении благоприятный.
Терапия кортикостероидами носит характер заместительной и обеспечивает нормальное развитие ребенка.
Профилактика — медико-генетическое консультирование.